Introduction
Since the first monoclonal antibody was approved by the U.S. Food and Drug Administration (FDA) in 1986, the market for therapeutic proteins has expanded rapidly, and antibody drugs have become predominant treatment modality for various diseases over the past 25 years 1). The stability of a drug molecule is very important because it affects the safety and efficacy of the drug. In products with higher order structures such as antibody drugs, the maintenance of molecular structure and biological activity depends on non-covalent bonds as well as covalent bonds of the molecule and is affected by various environmental factors 2).
In the guidelines of FDA and International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), the need for stability test data is described to understand how the quality of drug substances and drug products changes over time in different environments 3, 4). Therefore, the Forced Degradation Study (FDS) to purposely deteriorate and degrade the drugs is conducted at each stage from the early stage of research and development through marketing approval of antibody drugs. FDS is an effective method for clarifying the degradation pathways and structures of degradation products of drug substances and drug products, and guaranteeing stability. Also, it is useful for the development of manufacturing processes and selection of storage conditions and container 5, 6). Common test conditions for FDS include heating, freezing and thawing, agitation, pH, light irradiation, oxidation, reduction, and glycation 7, 8). Since one of the degradation pathways that occurs under many of these conditions is aggregates that are deeply related to immunogenicity, size exclusion chromatography (SEC) that can successfully separate and detect monomer and aggregates including multimers is used as an analytical method 5, 7).
In this article, we performed various forced degradation tests on MabThera® (rituximab), which is a therapeutic drug for non-Hodgkin’s lymphoma, and measured untreated and forcibly degraded MabThera® by SEC in order to compare the changes in the amount of monomer and aggregates including multimers and degradation products.
Experimental
<Instruments>
Pump: PU-4080i*
Autosampler: AS-4050i*
Column oven: CO-4060
UV detector: UV-4075*
* with option units
<Conditions>
Column: TSKgel G3000SWXL (7.8 mmI.D. x 300 mmL, 5 µm)
Eluent: 0.2 mol/L sodium phosphate buffer (pH 6.7)
Flow rate: 0.8 mL/min
Column temp.: 25 ºC
Wavelength: 220 nm
Inj. volume: 10 µL
Standard: Protein standard mix 15-600 kDa (Merck) 30 mg/mL in water
Sample: MabThera® 100 mg/10 mL (Roche) (Pretreatment methods are described later.)
Keywords
biopharmaceutical, antibody drug, FDS, aggregate, degradation product, MabThera®, TSKgel G3000SWXL, SEC, UV detector
Results
Figure 1 shows UV chromatograms of a blank (ultrapure water) and a protein standard sample. The column used in this experiment has exclusion limit with globular proteins of 800 kDa and measurement range of approximately 10 to 500 kDa. As shown in Figure 2, a molecular weight calibration curve was created from the retention times and molecular weights of the four components except for p-aminobenzoic acid that eluted after the permeation limit (around 13.8 min). Based on this calibration curve, the molecular weight of each sample was calculated.
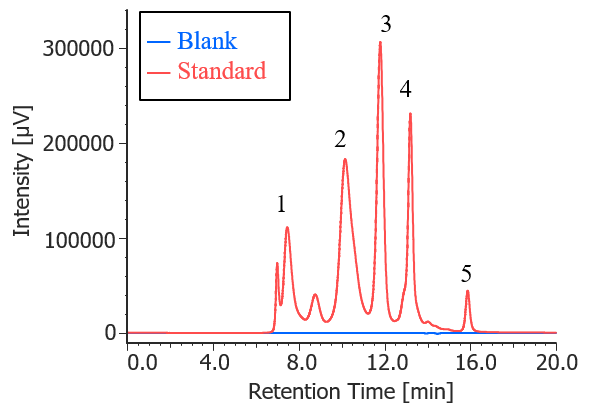
Fig. 1 UV chromatograms of a blank and a protein standard
(1: Thyrogrobulin (670 kDa), 2: γ-globulins (150 kDa), 3: Ovalbumin (44.3 kDa), 4: Ribonuclease A (13.7 kDa), 5: p-amino-benzoic acid (0.137 kDa))
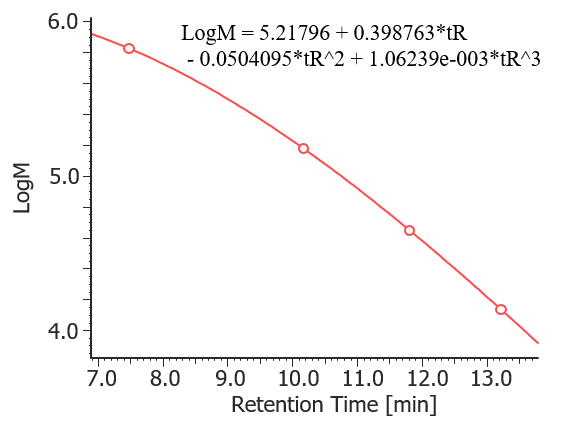
Fig. 2 Molecular weight calibration curve
Figure 3 shows the UV chromatogram of MabThera® without degradation treatment. Figures 4 to 7 show UV chromatograms of MabThera® that had been oxidized, glycated, deamidated, and heated, respectively. In addition, Figures 8 to 10 show the UV chromatograms of light-irradiated MabThera® with the irradiation time changed in three steps of 1, 2, and 5 minutes. The peaks around 14 min in Figures 3 to 10 are the peaks that eluted after the permeation limit of the column. Although detailed calculation results of molecular weight are not shown here, the monomer peaks were identified from each peak top molecular weight in Figures 3 to 10.
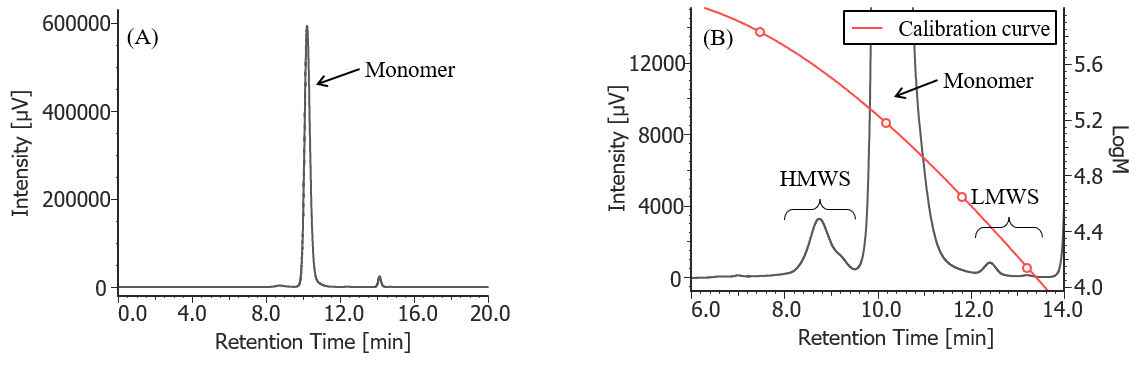
Fig. 3 UV chromatogram of untreated MabThera®
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with molecular weight calibration curve)
Pretreatment: 100 mg/10 mL of MabThera® was diluted with ultrapure water to 1 mg/mL.
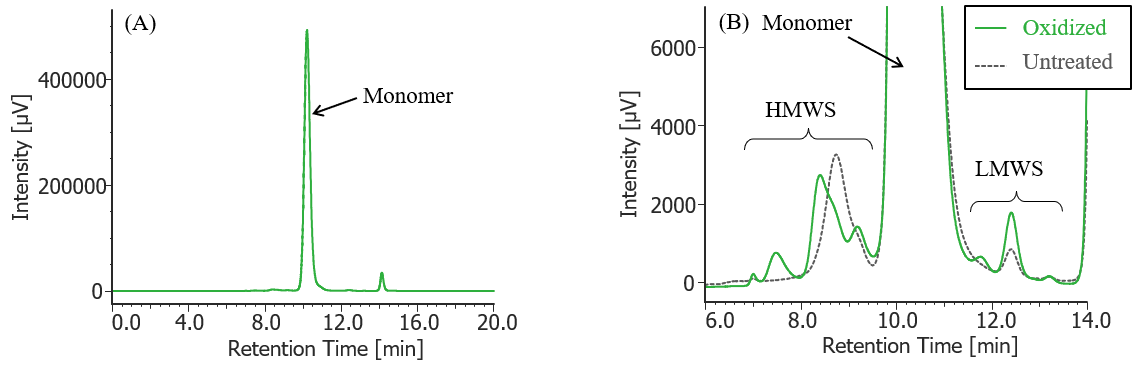
Fig. 4 UV chromatogram of oxidized MabThera®
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: One µL of 30% hydrogen peroxide solution was added (final concentration: 0.3% hydrogen peroxide) to 100 µL of MabThera® 100 mg/10 mL solution, and the solution was left at room temperature for 3 hours. Then, the solution was dialyzed against 10 mM sodium phosphate buffer (6 mM disodium hydrogen phosphate-4 mM potassium dihydrogen phosphate, pH 7.0) using a dialysis cellulose tube (Visking tubing, manufactured by Serva), and finally diluted 10 times with ultrapure water.
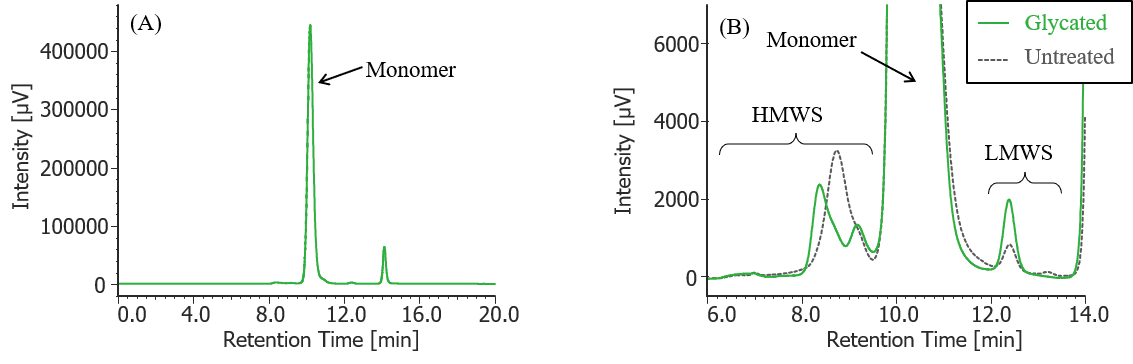
Fig. 5 UV chromatogram of glycated MabThera®
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: Fifty-four mg of glucose was added to 100 µL of MabThera® 100 mg/10 mL solution and dissolved (final concentration: 2.5 M glucose), and then left in a constant-temperature oven (SCF-Sro) at 40 ºC for 1 week. Then, the solution was dialyzed against 10 mM sodium phosphate buffer (6 mM disodium hydrogen phosphate-4 mM potassium dihydrogen phosphate, pH 7.0) using a dialysis cellulose tube (Visking tubing, manufactured by Serva), and finally diluted 10 times with ultrapure water.
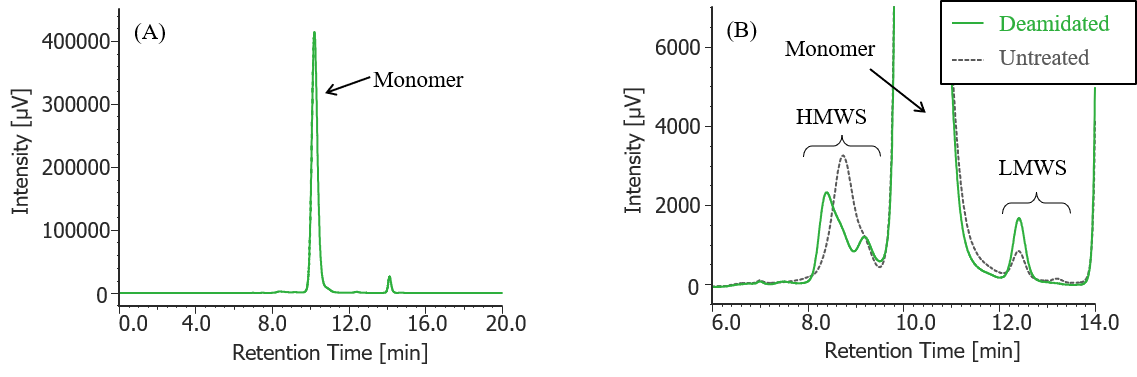
Fig. 6 UV chromatogram of deamidated MabThera®
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: 100 µL of MabThera® 100 mg/10 mL solution was dialyzed against 10 mM sodium phosphate buffer (6 mM disodium hydrogen phosphate-4 mM potassium dihydrogen phosphate, pH was adjusted to 8.5 by 5 M sodium hydroxide solution) using a dialysis cellulose tube (Visking tubing, manufactured by Serva), and then left in a constant-temperature oven (SCF-Sro) at 40 ºC for 1 week. Then, the solution was dialyzed against 10 mM sodium phosphate buffer (6 mM disodium hydrogen phosphate-4 mM potassium dihydrogen phosphate, pH 7.0) again, and finally diluted 10 times with ultrapure water.

Fig. 7 UV chromatogram of heated MabThera®
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: 100 µL of MabThera® 100 mg/10 mL solution was left in a constant-temperature oven (SCF-Sro) at 40 ºC for 8 days and then diluted 10 times with ultrapure water.
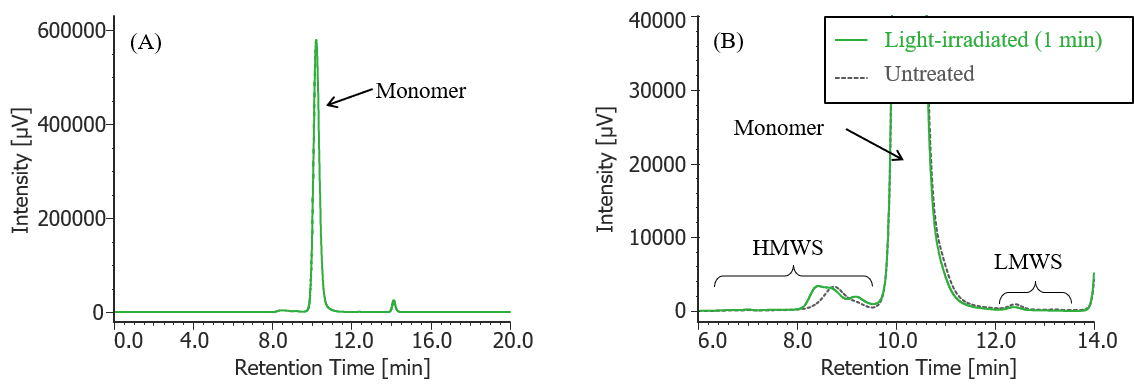
Fig. 8 UV chromatogram of light-irradiated MabThera® (irradiation time: 1 min)
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: 100 µL of MabThera® 100 mg/10 mL solution was irradiated with light (energy: 16.3 mW, center wavelength: 247 nm) for 1 minute using a light irradiation system (MM-3, manufactured by Bunkoukeiki).
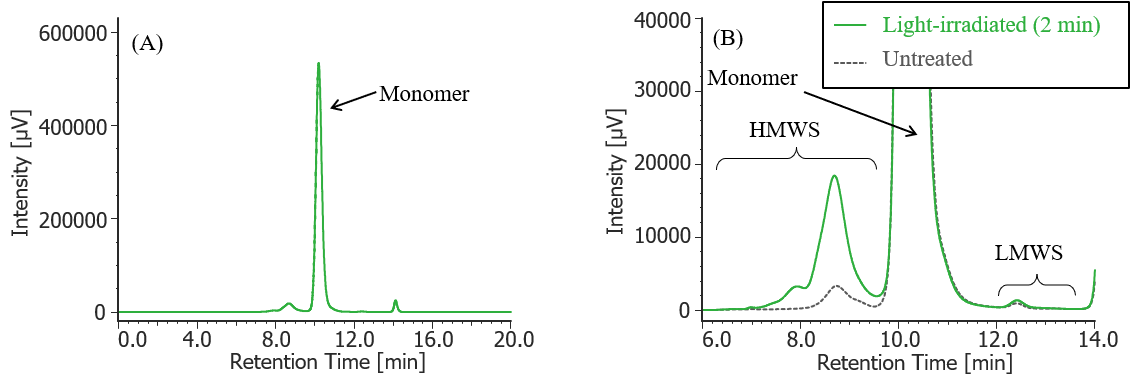
Fig. 9 UV chromatogram of light-irradiated MabThera® (irradiation time: 2 min)
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: 100 µL of MabThera® 100 mg/10 mL solution was irradiated with light (energy: 16.3 mW, center wavelength: 247 nm) for 2 minutes using a light irradiation system (MM-3, manufactured by Bunkoukeiki).
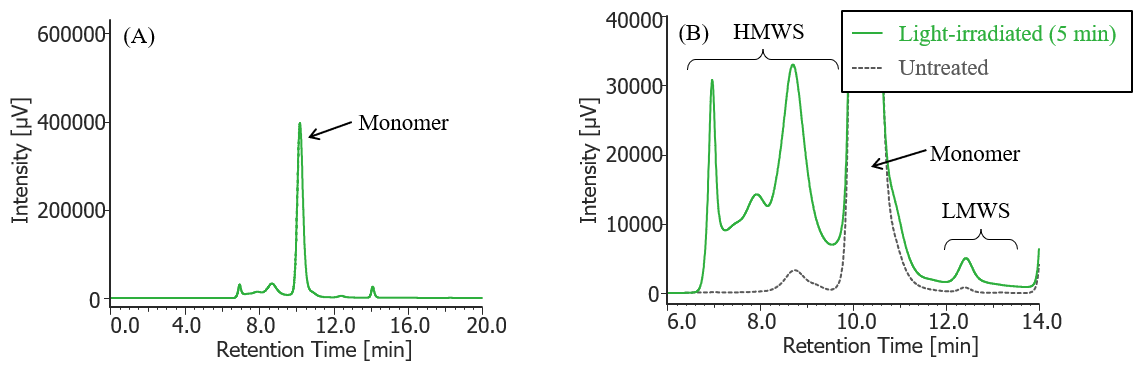
Fig. 10 UV chromatogram of light-irradiated MabThera® (irradiation time: 5 min)
(A) : Full scale, (B) : Enlarged view near the baseline at 6-14 min (shown with chromatogram of untreated MabThera® )
Pretreatment: 100 µL of MabThera® 100 mg/10 mL solution was irradiated with light (energy: 16.3 mW, center wavelength: 247 nm) for 5 minutes using a light irradiation system (MM-3, manufactured by Bunkoukeiki).
In Figures 3 to 10, the peaks on chromatograms were categorized into monomer, high molecular weight species (HMWS) including aggregates that elute earlier than monomer, and low molecular weight species (LMWS) including degradation products that elute later than monomer. Table 1 shows the comparison results of each category ratio (area %) to the total peak area. From these results the ratio of HWMS of MabThera® that had been oxidized, glycated, deamidated, heated, and light-irradiated (1min) increased by about 0.2-0.6% and that of LMWS increased by about 0.1-0.4% as compared to untreated MabThera®. Regarding light irradiation, it was found that the ratios of the monomer gradually decreased and those of the HMWS and LMWS increased significantly as the irradiation time increased.
Table 1 Comparison of ratios of HMWS, monomers, and LWMS in untreated and forcibly degraded MabThera®
| MW category | ||||
| HMWS | Monomer | LMWS | ||
| Type of treatments | Untreated | 1.0% | 98.8% | 0.2% |
| Oxidized | 1.5% | 97.9% | 0.6% | |
| Glycated | 1.2% | 98.3% | 0.5% | |
| Deamidated | 1.3% | 98.2% | 0.5% | |
| Heated | 1.6% | 98.1% | 0.3% | |
| Light-irritated (1 min) | 1.5% | 98.3% | 0.2% | |
| Light-irritated (2 min) | 6.7% | 93.0% | 0.3% | |
| Light-irritated (5 min) | 21.0% | 74.4% | 4.6% | |
Conclusion
In this experiment, it was confirmed that aggregates and degradation products in antibody drugs generated under various FDS test conditions can be well separated from monomers and detected by using SEC, and SEC is a powerful method for monitoring aggregates in the FDS.
References
1) R. M. Lu, Y. C. Hwang, I. J. Liu, C. C. Lee, H. Z. Tsai, H. J. Li, H. C. Wu, J. Biomed. Sci., 27 (1), 2 (2020). DOI: 10.1186/s12929-019-0592-z
2) European Medicines Agency: “ICH Topic Q5C Quality of Biotechnological Products: Stability Testing Biotechnological/Biological Products” <https://www.ema.europa.eu/en/documents/scientific-guideline/ich-topic-q-5-c-quality-biotechnological-products-stability-testing-biotechnological/biological-products_en.pdf>, (accessed 2022. 5. 30).
3) European Medicines Agency: “ICH Topic Q1A (R2) Stability Testing of new Drug Substances and Products” < https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf>, (accessed 2022. 5. 30).
4) U.S. Food and Drug Administration: “Guidance for Industry: INDs for Phase 2 and Phase 3 Studies” <https://www.fda.gov/media/70822/download>, (accessed 2022. 5. 30).
5) C. Nowak, J. K. Cheung, S. M. Dellatore, A. Katiyar, R. Bhat, J. Sun, G. Ponniah, A. Neill, B. Mason, A. Beck, H. Liu, mAbs, 9 (8), 1217 (2017). DOI: 10.1080/19420862.2017.1368602
6) Blessy M, R. D. Patal, P.N. Prajapati, Y. K. Agrawal, J. Pharm. Anal., 4 (3), 159 (2014). DOI: 10.1016/j.jpha.2013.09.003
7) J. Halley, Y. R. Chou, C. Cicchino, M. Huang, V. Sharma, N.C. Tan, S. Thakkar, L. L. Zhou, W. A. Azzam, S. Cornen, M. Gauden, Z. Gu, S. Kar, A. C. Lazar, P. Mehndiratta, J. Smith, Z. Sosic, P. Weisbach, E. S. E. Stokes, J. Pharm. Sci., 109, 6 (2020). DOI: 10.1016/j.xphs.2019.09.018
8) A. Hawe, M. Wiggenhorn, M. V. D. Weert, J. H. O. Garbe, H.C. Mahler, W. Jiskoot, J. Pharm. Sci., 101 (3), 895 (2012). DOI: 10.1002/jps.22812
Related Posts:
 In-situ Raman Monitoring for Flow Synthesis of Peptide Drugs
In-situ Raman Monitoring for Flow Synthesis of Peptide Drugs Streamlining Data Processing to Evaluate…
Streamlining Data Processing to Evaluate… Evaluation of Degradation of PET by Molecular Weight…
Evaluation of Degradation of PET by Molecular Weight… Evaluation of Degradation of POM by Molecular Weight…
Evaluation of Degradation of POM by Molecular Weight… Evaluation of Degradation of PA6 by Molecular Weight…
Evaluation of Degradation of PA6 by Molecular Weight… Degradation by Molecular Weight Distribution Measurements") Evaluation of Polystyrene (PS) Degradation by…
Evaluation of Polystyrene (PS) Degradation by…